Прионный белок PrPSc опаснее COVID-19

Пептиды в косметологии. Омолаживающий крем Liftiniti и Tisieye
25.11.2021
Тренды антивозрастных пептидов
02.12.2021
Прионные заболевания человека (трансмиссивные губчатые энцефалопатии) – это уникальные, прогрессирующие и смертельные нейродегенеративные заболевания, вызванные агрегацией в ткани нейронов неправильно сформированного прионного белка PrP. В связи с возможностью передачи TSE человеку в большинстве стран ведется активное наблюдение за распространением прионов – опасных белков, вызывающих заболевания со 100%-ной летальностью, что делает их более опасными, чем COVID-19. Рутинное использование диагностических критериев, включая биомаркеры, является достаточно надежным механизмом эпиднадзора. В обзоре рассмотрены типы инфекционных прионов, механизм действия, болезьни обусловленные ими и потенциальные методы лечения прионных заболеваний.
Общая характеристика белка PrP
Прионные болезни – это передающиеся, прогрессирующие и смертельные нейродегенеративные заболевания, связанные с агрегацией неправильно сформированного прионного белка (PrP) [1]. Трансмиссивные (т. е., передающиеся) губчатые энцефалопатии (TSE) человека включают болезнь Крейтцфельдта-Якоба (CJD), синдром Герстмана-Штраусслера-Шейнкера (GSS), куру и фатальную семейную бессонницу (FFI). Термин «прионы» был определен Стэнли Прусинером в 1982 году как «белковые инфекционные частицы» [2].
Естественный клеточный прионный белок PrPC имеет длину до 230 аминокислот и функционирует как гликолипидный сиалогликопротеин клеточной мембраны, локализованный в пресинаптических мембранах, который играет нейропротекторную и промиелинизирующую роль. Кроме того, он участвует в передачи нервных импульсов, транспорте цинка и меди, а также в гомеостазе кальция [1]. Более того, в лабораторных условиях PrPC способствует повышению устойчивости нейронов после ишемического инсульта головного мозга. Все прионные болезни человека связаны с патологической самовоспроизводящейся конформацией PrP, наиболее фундаментальной из которых является изменение третичной структуры PrP в результате посттрансляционных процессов в преобладающую β-листовую структуру. Этот процесс приводит к образованию заметно гидрофобной формы PrP с явной тенденцией к агрегации, последующей олигомеризации и образованию амилоидных фибрилл. Патологические агрегаты PrPSc (scrapie-изоформа прионного белка), возникающие в результате этого процесса, чрезвычайно устойчивы к физическим и химическим изменениям, и, в отличие от большинства белков, эти молекулы не денатурируются при кипячении. Прионные болезни человека определяются как передающиеся и быстро прогрессирующие дегенеративные заболевания центральной нервной системы, вызванные накоплением патологически конформированного PrP.
Исторически первые упоминания о болезни CJD относятся к 1920 и 1921 годам [3, 4], когда невролог Ганс Герхард Крейтцфельдт и невропатолог Альфонс Мария Якоб описали «нозологически очень тесно связанное, если не идентичное поражение» нескольких пациентов – куру. Самая последняя форма TSE, т. е., вариабельный синдром Крейцфельда-Якоба (vCJD), впервые выявлена в 1996 году в Великобритании [5] – BSE (Bovine spongiform encephalopathy, губчатая энцефалопатия КРС, т. н. «коровье бешенство») [6].
Образование прионов
Прионный белок, PrP, может принимать по меньшей мере две конформации: преобладающую клеточную конформацию (PrPC) и конформацию скрепи (PrPSc) [7]. Конформация PrPSc встречается очень редко и имеет характеристики амилоида. Кроме того, PrPSc является прионом, т. е., он заразен. Заражение происходит в два этапа: (1) PrPSc может шаблонировать PrPC и заставить его принять конформацию PrPSc и (2) PrPSc способен передаваться между людьми перорально, парентерально и другими путями и таким образом распространяться как инфекционный агент. Однако это упрощение: с одной стороны, PrPSc – это не единственная конформация, а скорее набор альтернативных похожих, но разных конформаций. Кроме того, существуют другие амилоидные конформации PrP с различными биохимическими и инфекционными свойствами.
Хотя PrP означает «прионный белок», сам PrPC не является прионом, но может быть превращен в прион. Действительно, прионы PrPSc распространяются путем шаблонирования своей особой конформации в PrPC, т. е., служат как «затравка» [8]. Это происходит через процесс, в котором образование водородных связей между амино- и карбонильными группами темплатирующего и темплатируемого полипептидов, вероятно, играет ключевую роль (Рисунок 1).

Рисунок 1 – Способность прионов к распространению и инфекционная природа. (1) В качестве ключевого механизма распространения прионов было предложено образование водородных связей между амино- и карбонильными группами разворачивающего и разворачиваемого полипептидов – таким образом PrPSc может распространяться по всему мозгу. (2) Инфекционный характер обусловлен тем, что PrPSc, введенный в другой мозг любым путем, может в нем распространяться. (PrP – прионный белок; PrPSc – прионный белок с конформацией скрепи).
Основной постулат теории прионов [2, 9] заключается в том, что при болезни клеточная форма PRNP, называемая PrPC, претерпевает конформационные изменения, в результате чего образуется «специфическая для скрепи» форма PrPSc. В свою очередь, PrPSc связывается с PrPC и способствует преобразованию PrPC→PrPsc, что приводит к усилению (предположительно нейротоксичного) PrPsc и развитию болезни
К настоящему времени каталог конформеров PrP значительно расширился. Структура PrPC хорошо известна [10]. Другие конфомеры менее изучены, но все они являются амилоидами и проявляют разные биохимические и биологические свойства.
Роль PRNP в организме
Интересно, что PRNP проявляет антимикробную активность, подавляя репликацию множества вирусов, а также взаимодействует непосредственно с пептидом амилоида-β (Aβ) болезни Альцгеймера (БА), чья антимикробная роль в настоящее время подтверждена [11]. PRNP и Aβ имеют общие мембранопроникающие, связывающие нуклеиновые кислоты и противовирусные свойства с классическими противомикробными пептидами, такими как LL-37.
PRNP повторяет механизмы агрегации и захвата патогенов, приписываемые Aβ и классическим антимикробным пептидам. Можно предположить, что преобразование PrPC в PrPSc может быть частью пути, параллельного преобразованию APP в Aβ и последующей агрегации, и активности по захвату патогенов, возможно, в прямой физической ассоциации с Aβ. Кроме того, для некоторых противомикробных пептидов связывание меди позволяет генерировать реактивные формы кислорода, которые способствуют инактивации связанных патогенов. Экспрессия PRNP повышается в результате инфекции [11], например, ВИЧ-1, аденовирусом 5, вирусами гепатита С, Эпштейна-Барр и Mycobacterium bovis, а также при воспалениях. Известны прямые доказательства того, что PRNP играет непосредственную защитную роль в защите от вирусной инфекции; можно с уверенностью заключить, что PRNP является защитной молекулой.
Прионные заболевания
Спорадические прионные заболевания человека
Болезнь Крейтцфельдта-Якоба (CJD) является наиболее распространенным прионным заболеванием человека [1]. Невропатологической характеристикой болезни Крейтцфельдта-Якоба является губчатая энцефалопатия в коре головного мозга и/или мозжечка и/или подкорковом сером веществе. В соответствии с различной этиологией выделяют три типа, наиболее распространенным из которых является спорадический (sCJD), генетический (gCJD) и приобретенный. Последний можно дополнительно разделить на ятрогенный (iCJD) и вариабельный (vCJD).
Спорадическая болезнь Крейтцфельдта-Якоба
Спорадическая болезнь Крейтцфельдта-Якоба начинается со случайного превращения физиологического PrPC в патологически конформированный PrPSc, что происходит примерно в 85% случаев [1] По имеющимся данным, заболеваемость синдромом Крейцфельда-Якоба в мире составляет от одного до двух случаев на миллион человек. В отличие от vCJD, клинические признаки и нейропатологические находки отличаются от случая к случаю, что, вероятно, обусловлено различными молекулярными фенотипами. Заболевание обычно длится, как правило, менее одного года. Лечения не существует, препараты могут лишь облегчить некоторые симптомы. Летальность составляет 100%.
Спорадическая фатальная бессонница
Спорадическая фатальная бессонница (sFI) определяется как быстро прогрессирующее нейродегенеративное заболевание с клиническим фенотипом, очень похожим на фатальную семейную бессонницу, характеризующееся неврологическим и когнитивным ухудшением наряду с тяжелыми нарушениями сна, преходящей диплопией и мозжечковой дисфункцией, с последующей утратой автономии, комой и 100%-ной смертью [1]. Лечения не существует.
Вариабельная протеазно-чувствительная прионопатия
Вариабельная протеаза-чувствительная прионопатия (VPSPr) – относительно недавно описанное прионное заболевание, выявленное в 2008 году [12]. VPSPr считается спорадической формой прионной болезни человека [1]. Средняя продолжительность заболевания составляет два года с клиническим преобладанием психиатрических признаков, афазии, атаксии и паркинсонического синдрома или когнитивного ухудшения. Невропатологически VPSPr характеризуется легкой губкообразной дегенерацией, в которой отсутствуют участки конфлюентной губкообразной трансформации, PrP-иммунореактивные «микробляшки», а также бляшкоподобные отложения. Лечения не существует, смертность 100%.
Приобретенная болезнь Крейтцфельдта-Якоба
Ятрогенная, случайно переданная iCJD
Ятрогенная форма возникает во время медицинских или хирургических процедур, в ходе которых происходит перенос патологически конформных прионов, и составляет менее 1% от всех случаев [1]. Передача прионов наблюдалась после пересадки твердой мозговой оболочки, от хирургических инструментов, после пересадки роговицы, глубокого введения электродов ЭЭГ, лечения гормоном роста человека и гонадотропинами. Инкубационный период сильно варьируется в зависимости от формы инокуляции. У пациентов с iCJD обычно наблюдаются аномалии походки и атаксия. Специальной терапии не известно.
Новый вариант болезни Крейцфельда-Якоба
Новый вариант болезни Крейцфельда-Якоба (vCJD) связан с употреблением продуктов, зараженных агентом BSE [1]. Клиническая картина вначале включает психиатрические и поведенческие симптомы, болезненные парестезии или дизестезию; атаксия и деменция развиваются позже. Продолжительность заболевания обычно 13–14 месяцев, а в нейропатологических результатах часто присутствуют мучнистые бляшки. Интересно, что известны три вероятные передачи инфекции через переливание крови от донора, страдающего vCJD; по этой причине существует запрет на использование доноров, живших в Великобритании во время эпидемии BSE. Болезнь смертельна.
Куру
Куру – нейродегенеративное, невоспалительное инфекционное заболевание [1]. Куру обнаружили около 1900 года в Папуа-Новой Гвинее среди каннибалистических племен, затем заболеваемость возросла в 1940–1950 годах. Мозжечковая атаксия, тремор и экстрапирамидные симптомы, такие как хорея и атетоз, были типичными симптомами для большинства пациентов, хотя результаты невропатологических исследований различались. Когнитивные нарушения отсутствовали. В нейропатологических отчетах описывается дегенерация миелина и нейронов, пролиферация микроглии и астроглии, мононуклеарная периваскулярная инфильтрация, манжетки, губчатая трансформация, уменьшенные нейроны с дисперсией вещества Ниссля с внутрицитоплазматическими вакуолями, вакуолизированные клетки Пуркинье мозжечка и нейроны стриатума. Амилоидные бляшки куру были зарегистрированы в 50–75% исследованных мозгов. В настоящее время болезнь куру считается исчезнувшей.
Наследственные прионные заболевания
Генетическая форма болезни Крейцфельда-Якоба
Генетическая/семейная форма обусловлена наличием наследственной мутации в гене PRNP, которая встречается в 10–15% случаев болезни CJD [1]. Клинически описывается деменция с другими психиатрическими изменениями, наряду с атаксией и миоклонусом, однако параличи взора и невропатии встречаются редко. Течение заболевания обычно более длительное, болезнь смертельна.
Синдром Герстмана-Штраусслера-Шейнкера
Синдром Герстмана-Штраусслера-Шейнкера (GSS) определяется как медленно прогрессирующее наследственное аутосомно-доминантное нейродегенеративное заболевание или энцефало(миело)патия с мультицентрическими бляшками PrP в коре головного и мозжечкового мозга и базальных ганглиях [1]. Именно мультицентрические бляшки являются невропатологической отличительной чертой GSS. GSS обычно проявляется мозжечковой атаксией и медленно прогрессирующей деменцией; тем не менее, экстрапирамидные симптомы, нарушения зрения и слуха, миоклонус, спастический парапарез, гипорефлексия или арефлексия в нижних конечностях также зарегистрированы как общие симптомы. Болезнь неизлечима.
Фатальная семейная бессонница
Фатальная семейная бессонница (FFI) -– аутосомно-доминантное наследственное заболевание, вызванное мутацией D178N в гене PRNP, связанной с наличием полиморфизма MM в кодоне 129 [1]. FFI характеризуется лекарственно-устойчивой бессонницей, фрагментацией сна, нарушениями вегетативной нервной системы, двигательными расстройствами и прогрессирующими когнитивными нарушениями. Наиболее пораженными областями являются медиодорсальное и переднее вентральное ядра таламуса, затем пульвинар и оливы. Обширная потеря нейронов и астроцитарный глиоз являются основными нейропатологическими находками, в то время как губкообразные трансформации отсутствуют. Лечения не существует, применение мощных снотворных дает лишь краткосрочное облегчение.
Передающиеся губчатые энцефалопатии животных и человека можно обобщить следующим образом (см. Рисунок 2).

Рисунок 2 – Передающиеся губчатые энцефалопатии
Лечение прионных заболеваний
В настоящее время не существует эффективных методов лечения прионных заболеваний [1, 13]. Терапия может быть направлена на различные мишени: PrPC, PrPTSE, преобразование PrPC в PrPTSE и конкретные пути нейродегенерации. На сегодняшний день большинство подходов основано на ингибировании накопления PrPTSE. Поскольку не существует досимптоматических тестов для диагностики прионных заболеваний, эти методы лечения назначаются на клинической стадии, после того как накопление PrPTSE уже произошло.
Одним из наиболее перспективных методов лечения прионных заболеваний человека является пассивная иммунизация анти-PRP антителами. Эта стратегия была распространена на несколько нейродегенеративных заболеваний, включая TSE, чему способствовало наблюдаемое снижение бремени амилоидных бляшек в трансгенной мышиной модели БА, обработанной антителами, направленными против Aβ.
Терапевтические методы, направленные на блокирование механизма, приводящего к образованию PrPTSE, являются хорошим подходом к лечению прионных заболеваний. Тем не менее практическое применение этих методов лечения зависит от разработки надежных диагностических тестов, позволяющих вводить антитела на ранней стадии заболевания. Активная иммунизация была оценена в ряде исследований с весьма обнадеживающими результатами
Заключение
Прионные болезни – это заболевания, крайне разрушительные для ЦНС. В настоящее время не существует эффективного лечения этих заболеваний, поэтому пациенты умирают в течение нескольких месяцев после появления первых клинических симптомов. Выявление нейротоксической молекулы и клеточных путей, ведущих к нейродегенерации, будет способствовать разработке новых методов лечения. Кроме того, создание ранних, неинвазивных диагностических тестов чрезвычайно важна как с эпидемиологической точки зрения, чтобы предотвратить вторичную передачу инфекции, так и с терапевтической, чтобы обеспечить применение эффективных методов лечения до того, как произойдет обширное повреждение мозга. Это особенно актуально в случае наследственных/генетических форм TSE.
Понимание биологии, лежащей в основе нейротоксичности, опосредованной прионами, является сложной задачей, учитывая беспрецедентную природу инфекционного агента. PrPTSE реплицируется путем придания своей конформации клеточному белку PrPC. Исследования показали, что потеря функции PrPC не может объяснить прионную нейротоксичность. В свете гетерогенности клинических и патологических проявлений разнообразной группы прионных заболеваний, весьма вероятно, что в формах TSE различной этиологии задействованы разные механизмы.
В целом, результаты свидетельствуют о том, что в последние десятилетия вышло множество работ по прионам (Рисунок 3) и достигнут значительный прогресс в выяснении механизмов вызванной ими нейродегенерации, и, что более важно, потенциальных методов лечения. На основе этих исследований можно будет разработать и внедрить в реальную жизнь соответствующие методы ранней диагностики и целенаправленного лечения людей и животных.

Рисунок 3 – Прионы в научных публикациях
1. Jankovska, N., Rusina, R., Bruzova, M., Parobkova, E., Olejar, T., Matej, R., Human Prion Disorders: Review of the Current Literature and a Twenty-Year Experience of the National Surveillance Center in the Czech Republic. Diagnostics, 2021. 11(10). DOI: 10.3390/diagnostics11101821.
2. Prusiner Stanley, B., Novel Proteinaceous Infectious Particles Cause Scrapie. Science, 1982. 216(4542): p. 136-144. DOI: 10.1126/science.6801762.
3. Creutzfeldt, H.G., Über eine eigenartige herdförmige erkrankung des zentralnervensystems (Vorläufige mitteilung). Zeitschrift für die gesamte Neurologie und Psychiatrie, 1920. 57(1): p. 1-18. DOI: 10.1007/BF02866081.
4. Jakob, A., Über eigenartige erkrankungen des zentralnervensystems mit bemerkenswertem anatomischen befunde. Zeitschrift für die gesamte Neurologie und Psychiatrie, 1921. 64(1): p. 147-228. DOI: 10.1007/BF02870932.
5. Will, R.G., Ironside, J.W., Zeidler, M., Estibeiro, K., Cousens, S.N., Smith, P.G., Alperovitch, A., Poser, S., Pocchiari, M., Hofman, A., A new variant of Creutzfeldt-Jakob disease in the UK. The Lancet, 1996. 347(9006): p. 921-925. DOI: 10.1016/S0140-6736(96)91412-9.
6. Brandel, J.-P., Vlaicu, M.B., Culeux, A., Belondrade, M., Bougard, D., Grznarova, K., Denouel, A., Plu, I., Bouaziz-Amar, E., Seilhean, D., Levasseur, M., Haïk, S., Variant Creutzfeldt–Jakob Disease Diagnosed 7.5 Years after Occupational Exposure. New England Journal of Medicine, 2020. 383(1): p. 83-85. DOI: 10.1056/NEJMc2000687.
7. Requena, J.R., The protean prion protein. PLOS Biology, 2020. 18(6): p. e3000754. DOI: 10.1371/journal.pbio.3000754.
8. Jarrett, J.T., Lansbury, P.T., Jr., Seeding «one-dimensional crystallization» of amyloid: A pathogenic mechanism in Alzheimer’s disease and scrapie? Cell, 1993. 73(6): p. 1055-1058. DOI: 10.1016/0092-8674(93)90635-4.
9. Prusiner, S.B., Gabizon, R., McKinley, M.P., On the biology of prions. Acta neuropathologica, 1987. 72(4): p. 299-314.
10. Prusiner, S.B., Prions. Proceedings of the National Academy of Sciences, 1998. 95(23): p. 13363-13383.
11. Lathe, R., Darlix, J.-L., Prion Protein PRNP: A New Player in Innate Immunity? The Aβ Connection. Journal of Alzheimer’s disease reports, 2017. 1(1): p. 263-275. DOI: 10.3233/ADR-170037.
12. Gambetti, P., Dong, Z., Yuan, J., Xiao, X., Zheng, M., Alshekhlee, A., Castellani, R., Cohen, M., Barria, M.A., Gonzalez-Romero, D., Belay, E.D., Schonberger, L.B., Marder, K., Harris, C., Burke, J.R., Montine, T., Wisniewski, T., Dickson, D.W., Soto, C., Hulette, C.M., Mastrianni, J.A., Kong, Q., Zou, W.-Q., A novel human disease with abnormal prion protein sensitive to protease. Annals of Neurology, 2008. 63(6): p. 697-708. DOI: https://doi.org/10.1002/ana.21420.
13. Saá, P., Harris, D.A., Cervenakova, L., Mechanisms of prion-induced neurodegeneration. Expert Reviews in Molecular Medicine, 2016. 18: p. e5. DOI: 10.1017/erm.2016.8.
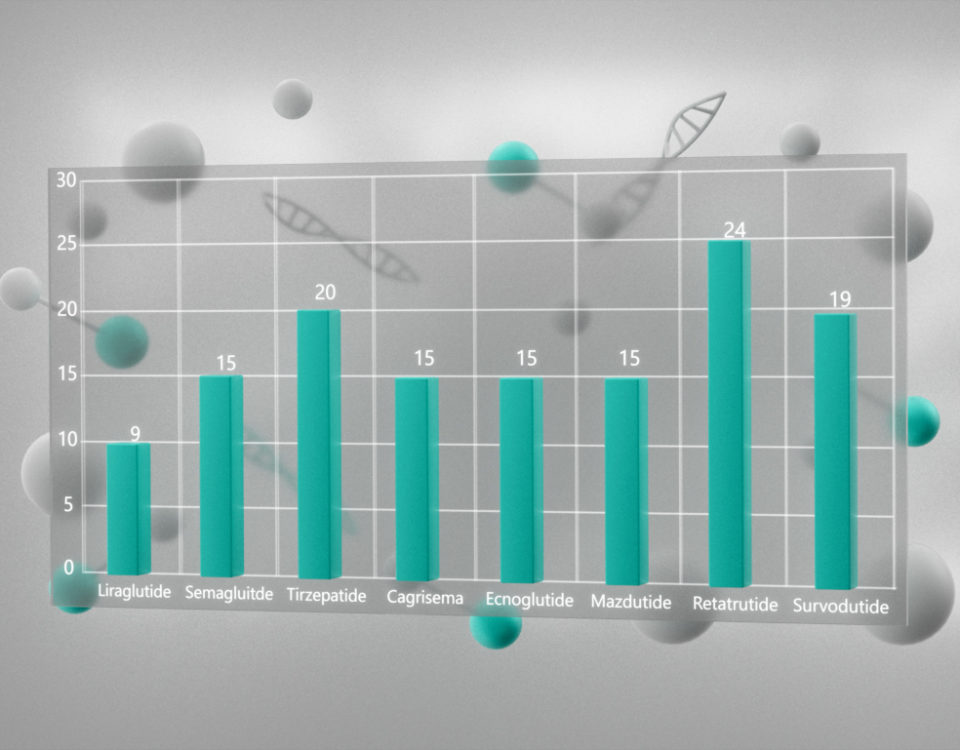


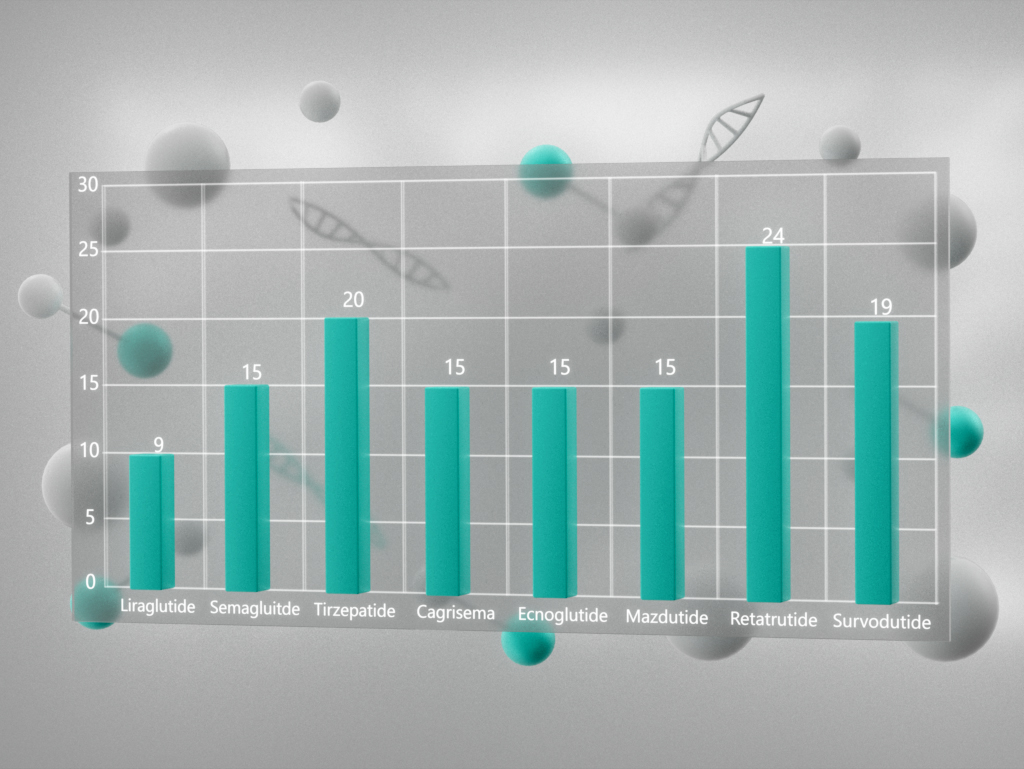{kind=link}
{kind=link}
{kind=link}
{kind=link}