
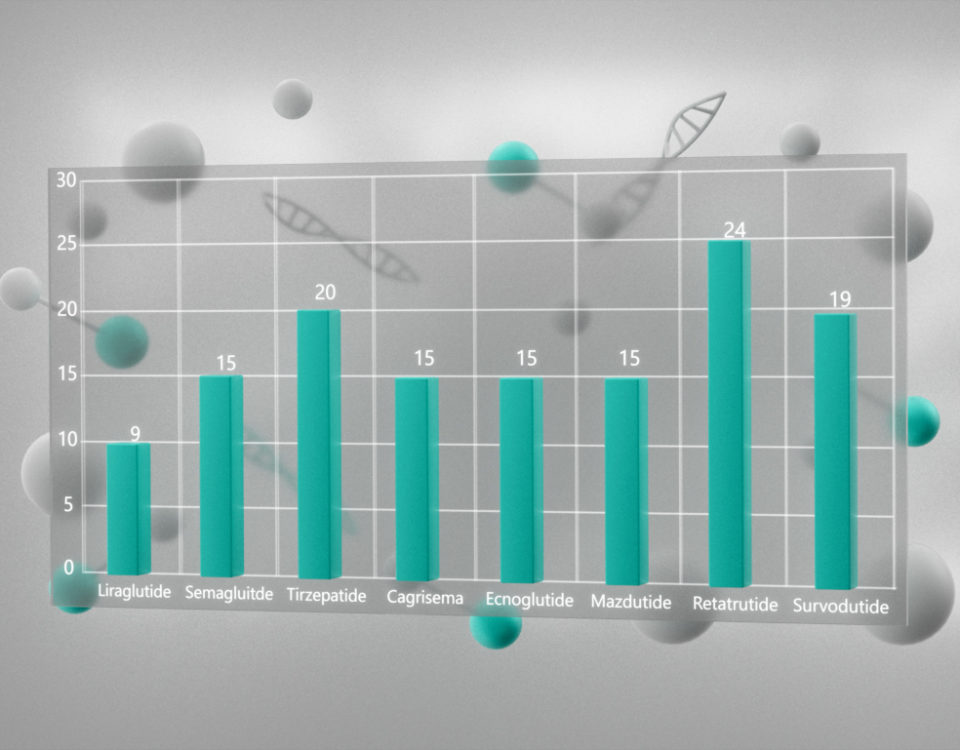
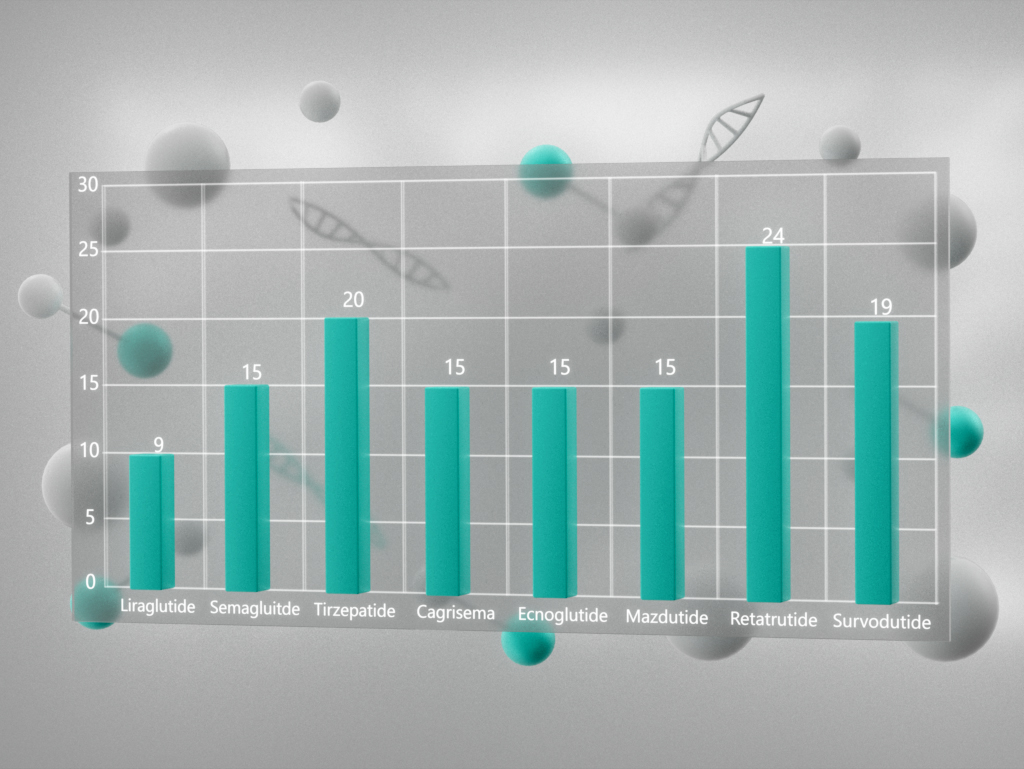
Топ три продаваемых пептидов в 2020 году: дулаглутид, лираглутид, семаглутид
16.04.2021
L-Аргинин. Либидо, сосуды, диабет, тревожность, спорт, старение, иммунитет
08.05.2021
К середине XX века наиболее распространённым методом синтеза пептидов был метод немецкого химика Эмиля Фишера, который первым начал работы в этой области. Метод Фишера получил название «классический синтез пептидов в растворе»
В 1963 году американский биохимик Мэррифилд публикует статью в American Chemical Society, в которой он описывает метод, названный им «твердофазный синтез пептидов». Эта статья стала одной из самых цитируемых публикаций в журнале. А в 1984 году Меррифилд становится лауреатом Нобелевской премии за выдающиеся достижения в области химического синтеза на твердой матрице, которая была названа «Смолой Меррифилда»
Твердофазный синтез пептидов – подход, совершивший революцию в органической химии и сделавший доступными сложные биологически активные молекулы. Его областями применения являются медицина, фармацевтика и биотехнология. Твердофазный синтез позволяет быстро получать продукты с высокой чистотой. В текущем обзоре рассмотрена история и основы концепции твердофазного синтеза пептидов и белков.
В твердофазном синтезе в качестве реагентов используются реакционноспособные фрагменты молекул, прикрепленных к нерастворимым полимерным подложкам. Наиболее широко этот подход применяется в синтезе полипептидов, полинуклеотидов, а также иммобилизованных ферментов и катализаторов. Отделение продуктов реакции от реагентов, примесей и катализаторов – это тот этап химического синтеза, на котором происходит много потерь. Дистилляция, перекристаллизация, хроматография и другие способы очистки никогда не бывают эффективными на 100%, а когда изолированные выходы меньше количественных, эти процессы являются причиной значительных потерь продукта, особенно в многостадийном синтезе. В твердофазном синтезе используется твердая смола, то есть полимерная подложка, к которой ковалентно прикреплены молекулы, а по завершению последовательных типовых реакций, позволяет легко отделить целевые вещества от реагентов.
История синтеза пептидов
Белки и пептиды играют важную роль в устройстве живых организмов. Они участвуют в большом количестве физиологических и биологических процессов, включая транспортировку веществ через мембраны, межклеточную коммуникацию и защиту. Проводится множество исследований, чтобы понять принципы и механизмы, управляющие функциональными и структурными характеристиками биоактивных белков и пептидов, которые, в свою очередь, могут быть использованы в медицинских, фармацевтических и биотехнологических приложениях. Такие работы требуют наличия белков и пептидов в разумных количествах и с максимальной чистотой. Белки и пептиды могут быть получены химическим синтезом, выделением из природных источников, рекомбинантными методами, используемыми при биохимической экспрессии белков, или комбинацией этих подходов.
Первый в истории синтез пептидов был выполнен Теодором Курциусом и Эмилем Фишером. Куртиус для докторской диссертации путем взаимодействия серебряной соли глицина с бензоилхлоридом синтезировал и охарактеризовал бензоилглицилглицин, первый N-защищенный дипептид. Эта работа была опубликована в 1882 году [1]. Девятнадцать лет спустя Эрнест Фурно и Эмиль Фишер [2] сообщили о синтезе глицилглицина, первого свободного дипептида, который был получен путем гидролиза дикетопиперазина. В 1902 году Фишер впервые ввел в обращение термин «пептиды».
Курциус разработал первый практический подход к синтезу пептидов с использованием реакции сочетания ацилазида. Фишер предложил другой метод с участием ацилхлорида, благодаря чему смог синтезировать пептиды, содержащие до 18 аминокислотных остатков. Оба метода имели некоторые ограничения, заключавшиеся в отсутствии легко удаляемых защитных групп для аминокислот.
Макс Бергманн и Леонидас Зервас в знаменательной публикации 1932 года [3] описали использование карбобензоксигруппы (Cbz, или «Z» – в честь Зерваса) в качестве временной защиты аминогруппы на N-конце, удаляемой путем каталитического гидрирования без воздействия на пептидную связь (удаление Z с серосодержащих остатков метионина и цистеина таким способом не удается из-за отравления катализатора). Помимо этого Z-группа, которая является группой уретанового типа, может быть удалена различными методами, включая ацидолиз с HBr и восстановление натрием в жидком аммиаке. Концепция временных защитных групп произвела революцию в представлениях химиков и привела к новой эре в синтезе пептидов.
В 1950-ых годах пептидный синтез превратился в зрелый подход, и были разработаны синтетические методы, которые позволили получать практически любой небольшой пептид. Например, в начале 1950-ых годов был синтезирован октапептид окситоцин, биологически и химически идентичный природному [4]. Акабори и сотрудники опубликовали один из первых асимметричных синтезов аминокислот [5]. В 1957 году Альбертсоном и Маккеем была введена трет-бутилоксикарбонильная (Boc) группа в качестве кислотнолабильной аминозащитной группы [6]. Важность защитной группы Boc заключается в ее ортогональности по отношению ко многим другим защитным группам, доступным для пептидного синтеза в то время. Понятие ортогональности будет раскрыто далее. Boc устойчив к сильным щелочам, восстановлению натрием и каталитическому гидрированию и, следовательно, полностью ортогонален Z- (или замещенным Z) группам. С этого момента многие пептиды были синтезированы с использованием комбинации Z- и Boc-защитных групп.
Основы пептидного синтеза
Для получения пептидов в основном применяются два подхода: один включает синтез пептидов на твердой фазе, а другой – в растворе, хотя в основном они оба основаны на одних и тех же принципах (Схема 1). Основное различие между двумя подходами заключается в природе защитной группы (PG1) карбоксильной группы первой аминокислоты. Когда PG1 является нерастворимой смолой, подход называется твердофазным пептидным синтезом, а когда PG1 растворим в реакционной среде, то жидкофазным.

Схема 1 – Принцип пептидного синтеза (PG1 – защитная группа COOH, PG2 защитная группа NH2)
Как показано на схеме 1, образование пептидной связи может быть достигнуто сочетанием двух аминокислотных остатков: один защищен на С-конце, а другой на N-конце. Если следовать стратегии жидкофазного синтеза, образованный защищенный дипептид затем подлежит выделению, очистке и характеристике. После удаления Nα-защитной группы (PG2) дипептидный амин затем связывается с другой аминокислотой, защищенной по Nα-аминогруппе, с получением защищенного трипептида, который при удалении превращается в трипептид-PG2 со свободной аминогруппой. Повторение конденсации и снятия защиты дает длинную защищенную пептидную цепь, с которой может быть полностью снята защита, давая конечный свободный пептид. Таким образом, для получения желаемого пептида, реакционные функциональные группы боковой цепи, Nα-амино- и карбоксильная, должны быть защищены соответствующим образом. Подход к синтезу пептидов в растворе трудозатратен и требует много времени, поскольку после каждого этапа должны выполняться выделение и очистка. Твердофазный синтез пептидов значительно упрощает сложные и длительные стадии очистки.
Твердофазный синтез пептидов и открытие Роберта Брюса Меррифилда
В 1963 году Меррифилд опубликовал первый в истории твердофазный синтез пептидов [7]. В исходной методике хлорметилированная нитрополистирольная смола была использована для синтеза тетрапептида Leu-Ala-Gly-Val (Схема 2а). Твердый носитель обрабатывали триэтиламмониевой солью Z-защищенного валина для образования бензиловой эфирной связи между Z-валином и твердым носителем (реакция SN2, Схема 2b). Связанный с полимером Z-валин промывали от избытка реагента и затем обрабатывали 30% HBr в уксусной кислоте для удаления Z-группы. Затем Z-защищенный глицин был присоединен к свободной аминогруппе валина посредством реакции с N,N’-дициклогексилкарбодиимид (DCC). И снова избыток реагентов удаляли промыванием c нерастворимого полимера. Цикл снятия защиты (удаление Z-группы), конденсации и промывки повторяли для последовательного добавления Z-AlaOH и Z-Leu-OH к растущей цепи с получением связанного с полимером тетрапептида. Обработка пептида на подложке гидроксидом натрия расщепила сложноэфирную связь с полимером, дав свободный тетрапептид Leu-Ala-Gly-Val.

Схема 2 – Оригинальный синтез Меррифилда
Позже Меррифилд усовершенствовал процедуру твердофазного синтеза пептидов [8], внеся в основную концепцию несколько важных модификаций, которые сократили время и улучшили общий выход. Boc-защищенные аминокислоты использовались вместо Z-защищенных. С этой модификацией не было необходимости использовать нитрованную смолу, а вместо этого применялась хлорметилсополистирол-дивинилбензольная. В 1984 году Меррифилд стал лауреатом Нобелевской премии за выдающиеся достижения в области химического синтеза на твердой подложке. В 1967 году Сакакибара представил HF-расщепление, с помощью которого можно удалять различные защитные группы [9]. Введение 9-фторенилметоксикарбонильной группы (Fmoc) в 1970 году Ханом и Карпино в качестве новой лабильной по отношению к основаниям защитной группы аминов [10], которая является ортогональной ко многим защитным группам, во многом способствовало распространению подхода Fmoc/t-Bu, в настоящее время являющегося наиболее часто используемой стратегией для твердофазного синтеза пептидов. За прошедшие годы процедуры были существенно улучшены, но основная идея осталась прежней.
Максимально возможная полнота конверсии достигается оптимизацией условий проведения реакций и использованием многократных избытков, 7–10 эквивалентов, реагентов и растворителей. Выбор смолы может повлиять на успех синтеза и чистоту сырья. Линкер определяет C-концевую функциональную группу в конечном продукте. Конденсирующий агент обычно зависит от требуемой скорости синтеза, при этом для быстрого требуются более высокореакционные (нестабильные) реагенты (HCTU, HATU и COMU), чем для медленного синтеза (DIC, HBTU). При этом, следует учитывать стерически затрудненные аминокислоты. Температура реакции может влиять на выбор реагента. Повышение температуры или применение микроволнового нагревания часто ускоряет синтез и улучшает чистоту, но не всегда. Для оптимизации протоколов важен мониторинг снятия защиты с помощью ультрафиолетового детектора в реальном времени. На заключительном этапе защитные Boc и Bzl группы должны быть удалены из целевых пептидов во время отщепления от подложки. Эти реакции включают гидролиз фтористым водородом (HF), и для улавливания карбокатионов, образующихся во время реакций SN1 вводят нуклеофилы (скавенджеры), например замещенные фенолы [11]. «Коктейли» с тиолами (EDT, DTT, DODT и другие) применяют при использовании Fmoc и отщеплении пептида с помощью TFA [12]. Это позволяет снизить загрязнение побочными продуктами окисления.
Смолы и линкеры в твердофазном синтезе пептидов
Концепция твердофазного синтеза пептидов основана на присоединении первой аминокислоты к смоле, а затем удлинении пептидной цепи для получения в конечном итоге целевого полипептида. Смола состоит из твердой полимерной основы, постоянно связанной с линкером (бифункциональный спейсер, промежуточный мостик или «рукоятка» – синонимы), который облегчает временное закрепление первой аминокислоты на твердой полимерной основе (Схема 3).

Схема 3 – Смола, линкер и пептид
Наиболее распространенным полимерным твердым носителем, используемым в смолах, является 1–2% дивинилбензол – сшитый полистирол в форме шариков диаметром около 50 микрон [13]. Имеющиеся в продаже смолы сшитого полистирола имеют обычно два размера частиц – 200–400 меш (35–75 мкм) и 100–200 меш (75–150 мкм). Существуют или разрабатываются альтернативные полимерные твердые подложки, включая, например, полиэтиленгликоль (PEG), полиэтиленгликоль-полистирольные смолы (PEG-PS), полиамидные подложки (обычные или заключенные в жесткие материалы, такие как полистирол с высокой степенью сшивки или кизельгур), бисерные сополимеры полиэтиленгликоля и акриламида (PEGA) и полиэтилен-полистирольные (PE–PS) пленки. Другие альтернативы рассмотрены в обзоре [14].
Твердые подложки из смол имеют большое значение, потому что синтез пептидов происходит внутри их набухшей трехмерной сетки, и большинство мест прикрепления находится внутри. В этом контексте важен фактор набухания, поскольку кинетика реакции контролируется диффузией. Как следствие, смола, которая набухает больше, будет иметь более высокую скорость диффузии реагентов в полимерную сетку, что приведет к быстрым реакциям и полноте химических превращений. Сшитые полистиролы нерастворимы в обычных растворителях, но они сольватируются и набухают в апротонных, таких как дихлорметан (DCM), N,N-диметилформамид (DMF) и тетрагидрофуран (THF). Добавление пептидов оказывает сильное влияние на сольватацию. По мере увеличения содержания пептида степень набухания в полярном ДМФА увеличивается. Противоположная тенденция наблюдается для неполярного растворителя DCM, где степень набухания уменьшается по мере увеличения количества связанных пептидных остатков.
Присутствие посторонних функциональных групп на смоле приводит к некоторым побочным реакциям, таким как трифторацетилирование и алкилирование сульфгидрильной группы цистеина п-гидроксибензильной группой, образуемой в результате разложения кислотного линкера Ванга. Побочные реакции также могут возникать либо из-за определенных последовательностей (образование аспартимида), либо из-за присутствия некоторых аминокислотных остатков (окисление метионина). Необходимо идентифицировать любую побочную реакцию, выяснить ее механизм и устранить причины. Многих побочных реакций можно избежать с помощью тщательного выбора синтетических протоколов, защитных групп боковых цепей и линкеров.
Хороший линкер должен удовлетворять ряду требований, включая способность иммобилизовать первый аминокислотный остаток с высоким выходом, стабильность ко всем химическим веществам, используемым в ходе твердофазного синтеза пептидов, и высокопродуктивное отделение целевого пептида без значительных побочных продуктов. Линкеры можно разделить на несколько категорий в соответствии с различными критериями, включая условия пептидного отщепления (фотолитическое, кислотное, основное, нуклеофильное замещение и т. д.) или тип функциональной группы, которая используется для иммобилизации первой аминокислоты на твердом носителе. Согласно последнему критерию, первая аминокислота может быть прикреплена к смоле через: 1) ее C-конец (стратегия C- к N-концу); 2) N-конец (стратегия N- к C-концу); 3) боковую цепь (если аминокислота имеет активную функциональную группу в боковой цепи); или 4) главную цепь (Схема 4a–d, соответственно).

Схема 4 – Стратегии иммобилизации пептидов
Концепция твердофазного синтеза пептидов от C— к N—концу, о которой впервые сообщил Меррифилд, по-прежнему остается преобладающей. Первая N-защищенная аминокислота иммобилизуется на смоле через ее карбоксильную группу. С-конец аминокислоты прикреплен к твердому носителю в виде амида, сложного эфира, сложного тиоэфира, О-замещенного оксима или гидразида (Схема 4a). Связанные со смолой сложные эфиры могут быть синтезированы взаимодействием N—защищенной аминокислоты либо с гидроксисмолой, либо с электрофильной, соответственно. С другой стороны, связанный смолой амид, гидразид, O—замещенный оксим или тиоэфир могут быть получены в результате реакции аминокислоты с амино-, гидразино-, оксимо- или сульфгидрильной смолой. После присоединения первой аминокислоты пептидная цепь может быть удлинена в направлении от C— к N—концу. Условия реакции для отщепления целевого пептида от смолы определяются типом линкера. При иммобилизации через С-конец используют стратегии Fmoc/tBu и Boc/Bzl. После отщепления пептида С-конец может стать карбоксильной или амидной группой, сложным эфиром, тиоэфиром, гидразидом, лактамом или лактоном.
Стратегия N— к C—концу основана на иммобилизации первой O-защищенной аминокислоты на смоле через ее аминогруппу и удлинении пептидной цепи путем добавления аминокислотных остатков от N— к C—концу (Схема 4b), и впервые описана Летсингером [15]. Она менее распространена и называется обратным твердофазным пептидным синтезом. Главное ограничение, связанное с этой стратегией, – потенциальная рацемизация через образование оксазолона. Кроме того, на каждой стадии также может образовываться дикетопиперазин. Наиболее часто используемые смолы для обратного твердофазного синтеза пептидов – силилхлоридная, имидазолидкарбаматная Ванга и 2-хлортритиловая. Связанный со смолой карбамат может быть получен взаимодействием О-защищенной аминокислоты с соответствующим линкером. Затем пептидная цепь может быть удлинена в обратном (N→C) направлении путем удаления временной защитной группы c карбоксильной, активации полученной свободной карбоксигруппы и последующей конденсации со следующей O-защищенной аминокислотой. Многократное повторение стадий снятия защиты, активации и конденсации дает пептид с желаемой длиной цепи. Затем целевой пептид может быть отщеплен от смолы в условиях, определяемых типом смолы.
Стратегия иммобилизации через боковую цепь требует подходящих заместителей в ней, а также C— иN-защиты первого аминокислотного остатка (Схема 4с). Следовательно, эту стратегию можно применять только к определенным аминокислотам: Glu, Asp (карбоксильная группа), Ser, Thr, Tyr, Hyp (гидроксильная группа), Arg (гуанидиногруппа), His, Trp, Lys, Orn (аминогруппа), Cys (сульфанильная группа) и Phe (фенильная группа). Смолы, используемые для закрепления боковых цепей, рассмотрены подробно в обзоре [16]. После присоединения первой аминокислоты твердофазный синтез можно проводить в обоих направлениях (C→N) и (N→C) с учетом селективного снятия защиты.
Другая вариация доступа к модифицированным по С-концу пептидам называется стратегией амидного линкера основной цепи (BAL – Backbone Amide Linker, Схема 4d). Стратегия BAL включает прикрепление первой аминокислоты к смоле через азот основной цепи. Смолы BAL содержат ароматические карбонильные линкеры, включая бензальдегид, ацетофенон, тиофен, нафталин, индол, фотолабильные альдегидные линкеры и защитные линкеры. Стратегия подразумевает иммобилизацию первойC-защищенной аминокислоты на смоле BAL путем восстановительного аминирования первичной аминогруппой карбонильной группы линкера с получением связанного со смолой вторичного амина, который затем ацилируется второй аминокислотой. Затем с аминогруппы второй аминокислоты снимается защита, и полученная свободная аминогруппа реагирует с активированной третьей аминокислотой с получением иммобилизованного трипептида. Затем пептидная цепь удлиняется путем повторения стадий снятия защиты и конденсации, за которыми следует, наконец, стадия отщепления. Различные применения стратегии BAL см. в [17]. Следует отметить, что отличием стратегии BAL от закрепления N-конца заключается в том, что в BAL закрепления аминогруппа первой аминокислоты присоединяется как к смоле, так и ко второй аминокислоте, что позволяет наращивать цепь в двух направлениях.
Конденсирующие агенты при постадийном удлинении пептидной цепи
Пептиды состоят из аминокислот, связанных амидными (пептидными) связями, следовательно, важнейшим этапом пептидного синтеза является образование таких связей. Получение амидной связи обработкой карбоновой кислоты амином нелегко, потому что они подвергаются кислотно-основной реакции с образованием соли аммония до того, как произойдет нуклеофильное замещение (Схема 5a).

Схема 5 – Взаимодействие кислот и аминов
Для решения этой проблемы добавляется дополнительный реагент –конденсирующий агент, чтобы активировать карбоксильную группу, превращая ОН в хорошую уходящую группой. Таким образом, карбоксильная группа активируется в направлении нуклеофильной атаки аминогруппой другого остатка. Для постадийного удлинения пептидной цепи используется широкий спектр реагентов. Условия реакции конденсации определяют скорость ацилирования, а также степень побочных реакций, включая рацемизацию. Приоритетом является поиск оптимального сочетания высокой скорости реакции и минимальной рацемизации. Например, конденсация в основных условиях может привести во время активации к катализируемой основанием рацемизации. Выявлены два основных пути рацемизации: 1) образование 5(4H)-оксазолона (Схема 6, путь A) и 2) прямая енолизация и образование энантиомерно нечистого активированного пептида (Схема 6, путь B). Далее основное внимание будет уделено наиболее часто используемым конденсирующим агентам.

Схема 6 – Рацемизация в синтезе пептидов
Карбодиимиды
N,N‘-Дициклогексилкарбодиимид (DCC) был впервые использован для синтеза пептидов в 1955 году [18]. DCC долго оставался самым популярным конденсирующим агентом как для твердофазного, так и жидкофазного подхода. Недостаток DCC заключается в образовании нерастворимого побочного продукта, симметричной мочевины, на стадии активации. Для решения этой проблемы рекомендуется использование N,N‘-диизопропилкарбодиимида (DIC). Также разработан N-этил—N‘(3-диметиламинопропил)карбодиимида гидрохлорид (EDC), известный как «водорастворимый карбодиимид» (WSC). EDC очень полезен для синтеза пептидов в жидкой фазе из-за хорошей растворимости его самого и соответствующей мочевины в водной среде, что позволяет легко удалять их при обработке. Два возможных пути образования пептидной связи посредством реакций активации и конденсации с карбодиимидами протекают либо через активную O-ацилизомочевину, либо симметричный ангидрид.
Активированные эфиры
Активированные сложные эфиры могут легко реагируют с рядом нуклеофилов, например с аминами. Такие эфиры обычно получают реакцией свободной карбоксильной группы либо с замещенным гидроксиламином, либо с замещенным фенолом, HO-Y в присутствии карбодиимида. Высоко реакционноспособный ацилирующий компонент затем реагирует со свободной аминогруппой, образуя амид. Электрофильность карбонильного углерода увеличивается за счет электроноакцепторных свойств Y-фрагмента сложного эфира. Обычно используемые реагенты сочетания HO-Y включают N-гидроксибензотриазол (HOBt) [19], N-гидроксисукцинимид (HOSu) и фенолы с электроноакцепторными заместителями – пентафторфенол, пентахлорфенол и п-нитрофенол. Следует отметить, что HOBt ускоряет аминолиз и подавляет рацемизацию, поэтому он широко используется в синтезе пептидов.
Соли фосфония
Использование солей фосфония в качестве конденсирующих агентов для образования амидной связи было впервые описано в 1969 году [20]. Соли фосфония получили широкое распространение для ускорения конденсации пептидов путем образования ацилфосфониевой соли после того, как был разработан (бензотриазол-1-илокси)трис(диметиламино)фосфония гексафторфосфат (BOP). С тех пор предложено множество солей фосфония, включая гексафторфосфаты бензотриазол-1-илокситри(пирролидино)фосфония (PyBOP), (6-хлорбензотриазол-1-илокси)трис(пирролидино)фосфония (PyClock), [(7-азабензотриазол-1-ил)окси]трис(пирролидино)фосфония (PyAOP), (((1-циано-2-этокси-2-оксоэтилиден)амино)окси)три(пирролидин-1-ил)фосфония (PyOxim или PyOxP) и бромтри(пирролидино)фосфония (PyBroP). Поскольку соли фосфония не реагируют со свободной Nα-аминогруппой, то могут быть добавлены непосредственно к аминогруппе входящего фрагмента и карбоксильному компоненту, который необходимо конденсировать, что означает, что эти реагенты могут давать активный сложный эфир in situ. С солями фосфония легко обращаться и они способствуют быстрому образованию пептидов. Предлагаемый механизм реакции включает промежуточные частицы, представляющие собой активный сложный эфир, в котором фосфор связан с атомом кислорода. Наконец, надо отметить, что недостатком использования BOP в качестве связывающего реагента является образование токсичного побочного продукта – гексаметилфосфоротриамида (HMPA). Поэтому рекомендуется избегать применения BOP.
Урониевые и аминиевые соли
Урониевые и аминиевые соли несут положительный заряд на атоме углерода, и используются в качестве конденсирующих агентов в синтезе пептидов. В 1978 году разработали и предложили для синтеза пептидов 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметиламиния гексафторфосфат (HBTU) [21]. HBTU и родственные соединения относятся к солям N-гуанидиния (аминия). На основе структуры HBTU создан ряд солей N-гуанидиния, включая 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетрафторборат тетраметиламиния (TBTU), 2-(6-хлор-1Н-бензотриазол-1-ил)-1,1,3,3-тетраметиламиния гексафторфосфат (HCTU) [22], 1-((диметиламино)(диметилимино)метил)-1H-[1,2,3]триазоло[4,5-b]пиридин-3-оксид агексафторфосфат (HATU) и 1- (пирролидин-1-иум-1-илиден(пирролидин-1-ил)метил)1H-[1,2,3]триазоло[4,5-b]пиридин-3-оксида гексафторфосфат (HAPyU). В сравнительных исследованиях было обнаружено, что HATU дает лучший выход конденсации при меньшей степени энантиомеризации, чем PyBOP, HBTU или TBTU. Были также разработаны родственные соли O-урония, включая 2-сукцинимидо-1,1,3,3-тетраметилурония тетрафторборат (TSTU), O-[(этоксикарбонил)цианометиленамино]-N,N,N‘,N‘-тетрафторборат тетраметилурония (TOTU) и (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламиноморфолинокарбения гексафторофосфат (COMU), который представляет собой новый реагент с эффективностью, сравнимой с HATU. В отличие от солей фосфония, аминиевые/урониевые реагенты имеют недостаток, заключающийся в их способности реагировать со свободной Nα-аминогруппой входящего фрагмента. Эта побочная реакция является проблематичной, поскольку гуанидинилирование свободной Nα-аминогруппы приводит к образованию укороченных пептидов, несущих N-концевую гуанидиногруппу. Следовательно, реагент нельзя добавлять непосредственно к свободной амино- и карбоксигруппам, подлежащим конденсации; это означает, что требуется предварительная активация карбоксильного компонента. Обычно ее проводят путем взаимодействия карбоксигруппы с конденсирующим агентом в надлежащих условиях, принимая во внимание, что он является ограничивающим реагентом, и тогда свободная Nα-аминогруппа входящего фрагмента добавляется к смеси для непосредственной реакции с активированным карбокси-компонентом.
Другие конденсирующие агенты, менее популярные в пептидном синтезе, включают симметричные и смешанные ангидриды, ацилазолы, ацилазиды, ацилгалогениды, фосфор- и сероорганические соединения, пиридиниевые производные и реагенты на полимерной основе.
Стратегия твердофазного пептидного синтеза Fmoc/t‑Bu и ортогональные защитные группы
Существует две стратегии получения пептидов: трет-бутилоксикарбонил/бензил (Boc/Bzl) и 9-фторенилметоксикарбонил/трет-бутил (Fmoc/t-Bu). В настоящее время предпочтительным является последний вариант твердофазного синтеза пептидов как для промышленных, так и для исследовательских целей. В стратегии Boc/Bzl защитной группой Nα-аминогруппы является Boc – временная группа, поскольку ее следует удалять на каждой стадии конденсации, тогда как защитные группы боковой цепи – постоянные, поскольку они остаются в течение всего синтеза и удаляются после завершения твердофазного синтеза. Каждое удаление временной защитной группы (Boc) достигается обработкой трифторуксусной кислотой (TFA, ТФУК). Повторяющийся ацидолиз с TFA для удаления Boc-группы может привести к побочным реакциям и изменению чувствительных пептидных связей. Заключительный этап, который подразумевает отщепление пептида от смолы и удаление постоянных защитных групп боковых цепей, происходит под действием сильной кислоты, такой как фтористый водород (HF), что опасно и требует специального лабораторного оборудования в виде тефлоновых резервуаров, малодоступных для многих исследовательских групп из-за дороговизны. Разработка стратегии Fmoc/t-Bu возникла из-за недостатков использования Boc/Bzl. В Fmoc/t-Bu Nα-аминогруппа защищена Fmoc. Удаление временной защитной группы (Fmoc) требует мягкой обработки основанием с использованием пиперидина, что в конечном итоге приводит к образованию желаемого свободного амино-компонента и образованию фульвен-пиперидинового аддукта в следствии реакции между дибензофульвеном и пиперидина (Схема 7).

Схема 7 – Удаление защитной группы Fmoc
Следует отметить, что группа Fmoc, оказалась непригодной для синтеза пептидов в жидкой фазе из-за ряда потенциальных побочных реакций между дибензофульвеном и высвобожденным амином. Fmoc нашла свое применение в результате разработки твердофазного синтеза пептидов с использованием полиамидных смол. Позже ее использовали с кизельгуровой смолой для синтеза с непрерывным потоком низкого давления. Смола на основе кизельгура состоит из сшитого и функционализированного полидиметилакриламидного геля, удерживаемого в порах твердых инертных макропористых неорганических частиц.
Защитные группы и аминокислоты
Аминокислоты можно разделить на три класса в зависимости от необходимости защиты боковой цепи: 1) защита не требуется для Ala, Gly, Leu, Ile, Val, Phe, Met и Pro; 2) защита является необязательной, но рекомендуется для His, Asn, Gln и Trp для решения потенциальных проблем, связанных с незащищенными боковыми цепями; и 3) защитные группы необходимы для Asp, Glu, Lys, Orn, Arg, Cys, Ser, Thr и Tyr. Защитные группы боковой цепи выбираются так, чтобы они были совместимы с выбранной временной защитной группой. Защита аминогрупп в боковой цепи Lys и Orn обычно достигается с использованием Boc для стратегии с использованием Fmoc и 2-хлорбензилоксикарбонила (2-Cl-Z) – с Boc.
Карбоксильные группы боковых цепей Asp и Glu обычно защищены в виде сложных эфиров. Для стратегии Fmoc в твердофазном синтезе пептидов используются t-Bu сложные эфиры, расщепляемые TFA-содержащими реагентами, тогда как для с Boc используются сложные бензиловые эфиры или более стерически затрудненные циклогексиловые, чтобы избежать некоторых побочных реакций, таких как циклизация Asp в аспартимид. Рекомендуемые защитные группы боковых цепей для стратегий Fmoc и Boc перечислены в работе [23] и обзоре [24].

Схема 8 – Стратегия Fmoc/t-Bu
На схеме 8 изображена стратегия Fmoc/t-Bu твердофазного синтеза пептидов, которая основана на последовательном добавлении Fmoc-защищенных аминокислотных остатков к нерастворимой смоле. Fmoc-защищенная аминокислота присоединяется к смоле с использованием подходящего конденсирующего агента с последующей промывкой иммобилизованного первого полупродукта N,N-диметилформамидом (DMF, ДМФА). Лабильная по отношению к основанию защитная группа Fmoc удаляется с использованием пиперидина с последующей промывкой DMF с образованием иммобилизованной аминокислоты с защищенной боковой цепью. Затем присоединяется вторая Fmoc-защищенная аминокислота с последующей стадией промывки и получением иммобилизованного дипептида. Затем циклы снятия защиты и конденсации повторяются много раз с получением иммобилизованного защищенного пептида, после чего с N-концевой аминокислоты удаляют Fmoc. Защитные группы боковых цепей часто выбирают так, чтобы они были ортогональны Fmoc и удалялись одновременно с отщеплением пептида от смолы. Снятие защиты с боковой цепи и отделение пептида обычно достигается обработкой TFA. На этой стадии полностью незащищенный пептид растворяется в смеси растворителей с TFA, и поэтому для осаждения пептида добавляют холодный диэтиловый эфир.
Успехи в твердофазном синтезе пептидов с помощью Fmoc сопровождаются применением ортогональной защитой. Это означает, что две функциональные группы могут быть защищены двумя разными защитными группами, а позже в синтезе одна из них может быть выборочно удалена, в то время как другая остается нетронутой, т. е., вторая функциональная группа остается защищенной. Стратегия ортогональной защиты позволяет получать большое количество важных синтетических пептидов, включая модифицированные и циклические биоактивные пептиды. На основе твердофазного синтеза пептидов были разработаны методы синтетической комбинаторной химии, с помощью которых синтезированы многочисленные пептидные библиотеки и внедрен одновременный параллельный синтез.
Соединение пептидных сегментов и образование амидных связей
Более пятидесяти лет развития пептидного синтеза сделали получение пептидных фрагментов в диапазоне 30–40 аминокислот обычными и рутинными делом. Для химического синтеза белков или более крупных пептидов может быть реализован один из двух подходов: конденсация пептидных сегментов, либо хемоселективное лигирование пептидов. Стратегия конденсации пептидных сегментов включает связывание в растворе защищенных пептидных фрагментов с последующим удалением всех защитных групп. С использованием этого подхода синтезирован ряд биологически релевантных белков и пептидов. Одной из крупнейших молекул, полученных таким способом, был предшественник зеленого флуоресцентного белка Aequorea (238 аминокислот) [25]. Хотя подход конденсации пептидных сегментов представляет собой мощную стратегию, он страдает рядом недостатков, которые не позволяют использовать его в качестве общего метода. Ограничения включают потенциальную эпимеризацию во время активации на С-конце пептидного сегмента, необходимость в больших количествах ценных пептидных сегментов, которые следует очищать на промежуточных стадиях, а также плохую растворимость защищенных боковых цепей сегментов.
Эти ограничения простимулировали разработку хемоселективного лигирования. Способы хемоселективного лигирования не требуют конденсирующих агентов или защиты боковой цепи. Большинство современных хемоселективных методов лигирования включают две этапа: вариабельный (обратимый) хемоселективный захват, за которым следует инвариантная (необратимая) перегруппировка. Поэтому два сокомпонента по лигированию должны иметь две функциональные группы X и Y, первая размещается на C-конце одного из реагентов пептидных сегментов, тогда как Y – размещен на N-конце другого. Группы X и Y уникальны и должны иметь возможность реагировать друг с другом хемоселективно и отличаться от многих других функциональных групп, присутствующих в системе. Для ковалентного связывания фрагментов разработан ряд методов хемоселективного лигирования. Наиболее распространенные из них приводят к следующим узлам: амиды (естественное химическое лигирование), лигирование KAHA (α-кетокислота–гидроксиламин) и лигирование серина/треонина, триазолы, фосфопрамидаты или оксимы. Методы хемоселективного лигирования рассмотрены в книгах [26, 27] и многих обзорах.
Заключение
Таким образом, спустя почти 60 лет после первого твердофазного пептидного синтеза, эта стратегия постоянно развивается и по-прежнему остается сложной и динамичной областью исследований. Необходимость получения пептидов и белков произвела революцию в представлениях химиков и биологов и привела к новой эре в биоорганической химии. Доступность чистых биоактивных пептидов и белков с помощью различных подходов к химическому синтезу позволила исследовать их функции in vivo и in vitro. Следовательно, это значительно облегчает исследования в области молекулярной биологии и биохимии за счет применения химически синтезированных и тщательно разработанных белков и пептидов. Несмотря на достижения в области полного химического синтеза полипептидов, все еще есть возможности для дальнейшего прогресса и улучшения существующих подходов.
Разработка твердофазного пептидного синтеза Меррифилдом открыла путь для автоматизированного синтеза – быстрого и удобного способа. Например, синтезатор AAPPTec Apex 396 имеет реакционные блоки для синтеза до 384 пептидов одновременно, включая реакции с нагревом и охлаждением (Рис. 1, слева), Intavis MultiPep RSi дает возможность синтеза на колонках (до 48 штук) или на планшетах (до 2х96 реакторных лунок) с подогревом и встряхиванием (Рис. 1, справа).

Рисунок 1 – Автоматические синтезаторы пептидов (из работы [28])
В будущем, остаются перспективными разработки новых линкеров и ортогональных защитных групп для более универсального и высокоэффективного твердофазного пептидного синтеза. Это облегчит получение сложных и длинных полипептидов в больших количествах. Применение усовершенствованных автоматических синтезаторов решит проблемы с воспроизводимостью и масштабируемостью реакций.
1. Curtius, T., Ueber einige neue der Hippursäure analog constituirte, synthetisch dargestellte Amidosäuren. Journal für Praktische Chemie, 1882. 26(1): p. 145-208. DOI: https://doi.org/10.1002/prac.18820260112.
2. Fischer, E., Fourneau, E., Über einige Derivate des Glykocolls, in Untersuchungen über Aminosäuren, Polypeptide und Proteïne (1899–1906), E. Fischer, Editor. 1906, Springer Berlin Heidelberg: Berlin, Heidelberg. p. 279-289.
3. Bergmann, M., Zervas, L., Über ein allgemeines Verfahren der Peptid-Synthese. Berichte der deutschen chemischen Gesellschaft (A and B Series), 1932. 65(7): p. 1192-1201. DOI: https://doi.org/10.1002/cber.19320650722.
4. Vigneaud, V.d., Ressler, C., Swan, C.J.M., Roberts, C.W., Katsoyannis, P.G., Gordon, S., The synthesis of an octapeptide amide with the hormonal activity of oxytocin Journal of the American Chemical Society, 1953. 75(19): p. 4879-4880. DOI: 10.1021/ja01115a553.
5. Akabori, S., Ikenaka, T., Matsumoto, K., On the Asymmetric Synthesis of Amino Acids. Proceedings of the Japan Academy, 1951. 27(1): p. 7-9. DOI: 10.2183/pjab1945.27.7.
6. McKay, F.C., Albertson, N.F., New Amine-masking Groups for Peptide Synthesis. Journal of the American Chemical Society, 1957. 79(17): p. 4686-4690. DOI: 10.1021/ja01574a029.
7. Merrifield, R.B., Solid Phase Peptide Synthesis. I. The Synthesis of a Tetrapeptide. Journal of the American Chemical Society, 1963. 85(14): p. 2149-2154. DOI: 10.1021/ja00897a025.
8. Merrifield, R.B., Solid-Phase Peptide Synthesis. III. An Improved Synthesis of Bradykinin*. Biochemistry, 1964. 3(9): p. 1385-1390. DOI: 10.1021/bi00897a032.
9. Sakakibara, S., Shimonishi, Y., Kishida, Y., Okada, M., Sugihara, H., Use of Anhydrous Hydrogen Fluoride in Peptide Synthesis. I. Behavior of Various Protective Groups in Anhydrous Hydrogen Fluoride. Bulletin of the Chemical Society of Japan, 1967. 40(9): p. 2164-2167. DOI: 10.1246/bcsj.40.2164.
10. Carpino, L.A., Han, G.Y., 9-Fluorenylmethoxycarbonyl function, a new base-sensitive amino-protecting group. Journal of the American Chemical Society, 1970. 92(19): p. 5748-5749. DOI: 10.1021/ja00722a043.
11. Samacá, J., Velandia-Bautista, E., Tabares, L., Escamilla, L., Vanegas, M., Patarroyo, M.-E., p-Methoxyphenol: A potent and effective scavenger for solid-phase peptide synthesis. Journal of Peptide Science, 2020. 26(6): p. e3251. DOI: https://doi.org/10.1002/psc.3251.
12. Pawlas, J., Svensson, T., Rasmussen, J.H., 1,4-Benzenedimethanethiol (1,4-BDMT) as a scavenger for greener peptide resin cleavages. RSC Advances, 2019. 9(67): p. 38928-38934. DOI: 10.1039/C9RA08553J.
13. Vaino, A.R., Janda, K.D., Solid-Phase Organic Synthesis: A Critical Understanding of the Resin. Journal of Combinatorial Chemistry, 2000. 2(6): p. 579-596. DOI: 10.1021/cc000046o.
14. Bergbreiter, D.E., Alternative polymer supports for organic chemistry. Medicinal Research Reviews, 1999. 19(5): p. 439-450. DOI: https://doi.org/10.1002/(SICI)1098-1128(199909)19:5<439::AID-MED9>3.0.CO;2-7.
15. Letsinger, R.L., Kornet, M.J., Popcorn Polymer as a Support in Multistep Syntheses. Journal of the American Chemical Society, 1963. 85(19): p. 3045-3046. DOI: 10.1021/ja00902a054.
16. Cherkupally, P., Acosta, G.A., Ramesh, S., De la Torre, B.G., Govender, T., Kruger, H.G., Albericio, F., Solid-phase peptide synthesis (SPPS), C-terminal vs. side-chain anchoring: a reality or a myth. Amino Acids, 2014. 46(8): p. 1827-1838. DOI: 10.1007/s00726-014-1746-7.
17. Boas, U., Brask, J., Jensen, K.J., Backbone Amide Linker in Solid-Phase Synthesis. Chemical Reviews, 2009. 109(5): p. 2092-2118. DOI: 10.1021/cr068206r.
18. Sheehan, J.C., Hess, G.P., A New Method of Forming Peptide Bonds. Journal of the American Chemical Society, 1955. 77(4): p. 1067-1068. DOI: 10.1021/ja01609a099.
19. König, W., Geiger, R., Eine neue Methode zur Synthese von Peptiden: Aktivierung der Carboxylgruppe mit Dicyclohexylcarbodiimid unter Zusatz von 1-Hydroxy-benzotriazolen. Chemische Berichte, 1970. 103(3): p. 788-798. DOI: https://doi.org/10.1002/cber.19701030319.
20. Gawne, G., Kenner, G.W., Sheppard, R.C., Acyloxyphosphonium salts as acylating agents. Synthesis of peptides. Journal of the American Chemical Society, 1969. 91(20): p. 5669-5671. DOI: 10.1021/ja01048a057.
21. Dourtoglou, V., Ziegler, J.-C., Gross, B., L’hexafluorophosphate de O-benzotriazolyl-N,N-tetramethyluronium: Un reactif de couplage peptidique nouveau et efficace. Tetrahedron Letters, 1978. 19(15): p. 1269-1272. DOI: https://doi.org/10.1016/0040-4039(78)80103-8.
22. Marder, O., Shvo, Y., Albiricio, F., HCTU and TCTU. New coupling reagents: development and industrial aspects. Chimica oggi : International journal of chemistry and biotechnology = Chemistry today, 2002. 20(7-8): p. 37-41.
23. Soural, M., Hlaváč, J., Krchňák, V., Linkers for Solid-Phase Peptide Synthesis, in Amino Acids, Peptides and Proteins in Organic Chemistry. p. 273-312.
24. Isidro-Llobet, A., Álvarez, M., Albericio, F., Amino Acid-Protecting Groups. Chemical Reviews, 2009. 109(6): p. 2455-2504. DOI: 10.1021/cr800323s.
25. Nishiuchi, Y., Inui, T., Nishio, H., Bódi, J., Kimura, T., Tsuji, F.I., Sakakibara, S., Chemical synthesis of the precursor molecule of the <em>Aequorea</em> green fluorescent protein, subsequent folding, and development of fluorescence. Proceedings of the National Academy of Sciences, 1998. 95(23): p. 13549. DOI: 10.1073/pnas.95.23.13549.
26. D’Andrea, L.D., Romanelli, A., Chemical ligation: Tools for biomolecule synthesis and modification. 2017: John Wiley & Sons.
27. Algar, W.R., Dawson, P., Medintz, I.L., Chemoselective and Bioorthogonal Ligation Reactions. 2017: Wiley Online Library.
28. Winkler, D.F.H., Automated Solid-Phase Peptide Synthesis, in Peptide Synthesis: Methods and Protocols, W.M. Hussein, Skwarczynski, M., Toth, I., Editors. 2020, Springer US: New York, NY. p. 59-94.




{kind=link}
{kind=link}
{kind=link}
{kind=link}